af Catherine Gara
Januar 2016—slim er måske ikke noget, vi kan lide at tænke på, men vores liv afhænger af det. Ved cystisk fibrose (CF) fører fortykkelsen af slim til lungeinfektioner og tarmblokke, blandt andre symptomer. Sygdommen er forårsaget af et enkelt gen, der er påvirket af en eller flere af de mere end 1.700 mutationer, der vides at forårsage lidelsen. Hos Johns Hopkins, forskere og kliniker genetikere arbejder sammen om at lære alt, hvad de kan om genet, dets protein og hvad der går galt hos patienter, vel vidende, at hvert stykke information, de samler, bringer dem tættere på at have behandlingsmuligheder for alle personer med CF.
Giv mig saltet
i disse dage er patienter med CF meget bedre stillet takket være behandlinger, der styrer deres hyppige lungeinfektioner, men en diagnose af CF betyder stadig en forventet levetid på kun 38 på grund af den vejafgift, det tager på bugspytkirtlen, leveren og tarmene. Problemet kommer fra defekter i cystisk fibrose transmembran konduktansregulator (CFTR) gen, som bærer planen for CFTR-proteinet. CFTR-proteinet er som postsporet i en hoveddør. Det danner en lille passage mellem indersiden og ydersiden af cellen.
når CFTR fungerer korrekt, hjælper det med at kontrollere passagen af chloridioner (en komponent af salt) ind og ud af cellen. I lungerne og kanalerne i bugspytkirtlen, når chlorid forlader celler, tilskynder det vand til at følge. Det vand hjælper med at danne tynde lag af slim. I lungerne fælder slimet støv og bakterier, der ikke burde være der. Cilia, eller hårlignende strukturer, på cellerne, der linjer lungerne, hyrder derefter slimet op ad luftvejen til munden, hvor det bliver slugt og sendt for at blive fordøjet. I bugspytkirtlen hjælper væsken med at bære tarm for at hjælpe med fordøjelsen af mad. Hvis slimet er for tyktflydende i lungerne, kan cilia ikke flytte det ud, så bakterier forbliver der for at forårsage infektioner; hvis det er for tyktflydende i bugspytkirtelkanalerne, når ikke tarmene, og mad fordøjes ikke ordentligt.Garry Cutting, professor ved Institut for genetisk medicin, og Bill Guggino, direktør for Institut for fysiologi, har studeret CFTR-genet og dets kodede protein i de fleste af deres karriere. Cutting interesse stammer fra omsorg for et par brødre med CF, mens en beboer på Johns Hopkins. Gugginos interesse går endnu længere tilbage-til hans drengeårsture til havet. Han spekulerede på, hvordan fisk kunne overleve i saltvand, og han lærte, at det korte svar er: deres version af CFTR.
hvis det synes svært at forestille sig at fokusere en hel karriere på et enkelt gen og dets proteinprodukt, tænk på det i stedet som en kompleks Rubiks terning lavet af en kæde af 1.480 magnetiske blokke (aminosyrer). En mutation i CFTR-genet vil ofte betyde en ændring i en af aminosyrerne, som dramatisk kan påvirke den endelige tredimensionelle form.
nogle mutationer får proteinet til ikke at blive lavet. Andre tillader, at et partielt protein syntetiseres. Jo tættere på proteinets begyndelse disse mutationer forekommer, jo værre for dets funktion. Andre mutationer forekommer på det forkerte sted og forhindrer for eksempel saltkanalen i at åbne. Atter andre gør CFTR fold forkert, hvilket signalerer cellens kvalitetskontrolteam for at hente det og genbruge det. Og andre forstyrrer ikke proteinets funktion, men de forhindrer det i at komme til plasmamembranen, hvor det gør sit arbejde.
Skæring og Gugginos indsats har bidraget til designet af to CF-lægemidler på markedet: ivacaftor og lumacaftor. Ivacaftor aktiverer CFTR, der bærer mutationen G551D. Cutting ‘ s lab rapporterede først denne mutation i 1990, og Guggino og Cutting genererede efterfølgende ny indsigt i effekten af denne mutation på CFTR-funktion og patienters symptomer. Desværre findes G551D-mutationen kun hos 4 procent af patienterne med CF. Det viser sig imidlertid, at 50 procent af patienterne med CF har to kopier af en anden mutation (kaldet delta F508), hvilket får CFTR til at blive dårligt dannet og sendt til cellens genbrugsbeholder. Lumacaftor forhindrer det i at blive genbrugt, så det gør det til plasmamembranen. Derefter giver ivacaftor det “spark”, det har brug for for at arbejde.”delta F508 CFTR er stadig handicappet, men det er bedre end ingenting,” siger Guggino. “Og det betyder, at vi ikke bare behandler symptomer længere. Vi behandler de grundlæggende årsager.”
intet Barn efterladt
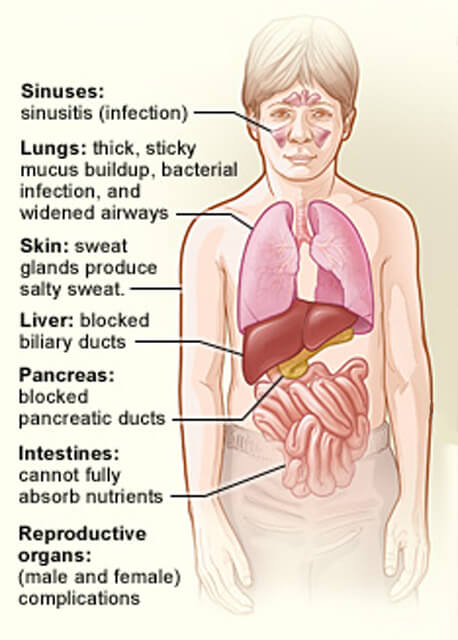 organerne påvirket af cystisk fibrose.
organerne påvirket af cystisk fibrose.kredit: National Heart, Lung and Blood Institute
de mere end 40 procent af patienter med CF med andre mutationer — nogle ret sjældne — er ikke altid så heldige. Der er over 1.700 mutationer i CFTR-genet, der forårsager CF, og kun en mindre fraktion er blevet testet for respons på de godkendte lægemidler. Nogle af de testede reagerer på det ene eller det andet stof, men andre gør det ikke. alligevel håber forskerne, at de en dag vil kunne hjælpe alle. Skæring forudser til sidst kategorisering af hver patient efter theratype, et ord han opfandt for at beskrive grupper af patienter, der sandsynligvis vil reagere på den samme terapi på grund af en almindelig underliggende årsag til deres symptomer. Han har arbejdet tæt sammen med kollegerne Patrick Sosnay og Karen Raraigh for at mine data fra 88.000 individer over hele verden for at teste hans koncept.
Vi kan bruge information genereret af eksperimenter i celler til at gruppere mutationer, der påvirker den samme egenskab af CFTR og derfor bør reagere på det samme panel af CFTR-lægemidler. Gruppering af mutationer efter theratype ville muliggøre kliniske forsøg på patienter, der bærer forskellige mutationer, i stedet for kliniske forsøg, der evaluerer en mutation ad gangen,” siger Cutting. “Det er præcisionsmedicin, der bliver en realitet.”
desværre virker lægemiddelbaserede terapier ikke for alle patienter med CF, især de 2 procent, der slet ikke laver CFTR. For disse udvikler forskere måder at målrette mod det muterede gen selv, selvom der stadig er mange forhindringer at overvinde.
Guggino har udarbejdet et genterapisystem, der bruger en modificeret adeno-associeret virus (AAV) til at deponere en god version af CFTR-genet inde i celler. Systemet har vist sig i humane luftvejsceller og gnavere.
forsøger en anden tilgang, Liudmila Cebotaru, fra Institut for medicin, udtænkte en ny måde at kombinere genterapi og proteinreparation ved hjælp af en mekanisme kaldet transcomplementation. I stedet for at placere CFTR-genet i fuld længde i AAV, bruger hun en kortere version, der lettere indsættes i cellens genom. Når det kortere protein produceres, binder det sig til patientens mutante protein og hjælper det med at komme til plasmamembranen. Både Cebotaru og Guggino tester nu sin nye tilgang hos rhesusaber, fordi deres lunger og immunsystem er meget tæt på mennesker. De håber at starte et klinisk forsøg i de næste par år, hvis alt går godt.
“Jeg kan godt lide at tænke på det som at hoppe på din bils batteri,” siger hun. “Med lidt ekstra hjælp kan patienternes CFTR-proteiner komme til deres destination.”
for både forskere og patienter er destinationen intet mindre end en kur mod CF. Og selvom det stadig er langt væk, der er opmuntrende tegn på, at vi kan komme dertil.