Di Catherine Gara
Gennaio 2016—Il muco potrebbe non essere qualcosa a cui ci piace pensare, ma le nostre vite dipendono da esso. Nella fibrosi cistica (CF), l’ispessimento del muco porta a infezioni polmonari e blocchi intestinali, tra gli altri sintomi. La malattia è causata da un singolo gene affetto da una o più delle oltre 1.700 mutazioni note per causare il disturbo. Alla Johns Hopkins, ricercatori e genetisti clinici stanno lavorando insieme per imparare tutto il possibile sul gene, la sua proteina e cosa va storto nei pazienti, sapendo che ogni informazione che raccolgono li sta avvicinando ad avere opzioni di trattamento per tutti gli individui con CF.
Passami il sale
In questi giorni, i pazienti con CF stanno molto meglio grazie a trattamenti che gestiscono le loro frequenti infezioni polmonari, ma una diagnosi di CF significa ancora un’aspettativa di vita di soli 38 a causa del pedaggio che assume su pancreas, fegato e intestino. Il problema deriva da difetti nel gene del regolatore di conduttanza transmembrana della fibrosi cistica (CFTR), che porta il progetto per la proteina CFTR. La proteina CFTR è come lo slot di posta in una porta d’ingresso. Forma un piccolo passaggio tra l’interno e l’esterno della cella.
Quando funziona correttamente, CFTR aiuta a controllare il passaggio di ioni cloruro (un componente del sale) dentro e fuori dalla cellula. Nei polmoni e nei dotti del pancreas, quando il cloruro lascia le cellule, incoraggia l’acqua a seguire. Quell’acqua aiuta a formare sottili strati di muco. Nei polmoni, il muco intrappola polvere e batteri che non dovrebbero essere lì. Le ciglia, o strutture simili a capelli, sulle cellule che rivestono i polmoni poi pastore il muco fino alle vie aeree fino alla bocca, dove viene ingerito e inviato per essere digerito. Nel pancreas, il fluido aiuta a trasportare gli enzimi nell’intestino per aiutare nella digestione degli alimenti. Se il muco è troppo viscoso nei polmoni, le ciglia non possono spostarlo, quindi i batteri rimangono lì per causare infezioni; se è troppo viscoso nei dotti pancreatici, gli enzimi non raggiungono l’intestino e il cibo non digerisce correttamente.
Percorso di una proteina
Garry Cutting, professore presso l’Istituto di Medicina Genetica, e Bill Guggino, direttore del Dipartimento di Fisiologia, hanno studiato il gene CFTR e la sua proteina codificata per la maggior parte della loro carriera. L’interesse di taglio deriva dalla cura di una coppia di fratelli con CF mentre un residente a Johns Hopkins. L’interesse di Guggino risale ancora più lontano – ai suoi viaggi di fanciullezza al mare. Si chiese come i pesci potrebbero sopravvivere in acqua salata, e ha imparato che la risposta breve è: la loro versione di CFTR.
Se sembra difficile immaginare di concentrare un’intera carriera su un singolo gene e il suo prodotto proteico, pensalo invece come un cubo di Rubik complesso costituito da una catena di 1.480 blocchi magnetici (amminoacidi). Una mutazione nel gene CFTR significherà spesso un cambiamento in uno degli amminoacidi, che può influenzare drammaticamente la forma tridimensionale finale.
Alcune mutazioni fanno sì che la proteina non venga prodotta. Altri permettono di sintetizzare una proteina parziale. Più vicino all’inizio della proteina si verificano queste mutazioni, peggio per la sua funzione. Altre mutazioni si verificano nel posto sbagliato e impediscono l’apertura del canale del sale, per esempio. Altri ancora fanno CFTR fold in modo improprio, che segnala il team di controllo qualità della cella per raccoglierlo e riciclarlo. E altri non interferiscono con la funzione della proteina, ma le impediscono di raggiungere la membrana plasmatica dove svolge il suo lavoro.
Gli sforzi di Cutting e Guggino hanno contribuito alla progettazione di due farmaci CF sul mercato: ivacaftor e lumacaftor. Ivacaftor attiva CFTR portando la mutazione G551D. Il laboratorio di Cutting ha riportato per la prima volta questa mutazione nel 1990, e Guggino e Cutting hanno successivamente generato nuove intuizioni sull’effetto di questa mutazione sulla funzione CFTR e sui sintomi dei pazienti. Sfortunatamente, la mutazione G551D si trova solo nel 4% dei pazienti con CF. Tuttavia, si scopre che il 50 per cento dei pazienti con CF hanno due copie di una mutazione diversa (chiamato delta F508), che causa CFTR per essere mal formato e inviato al cestino della cella. Lumacaftor impedisce che venga riciclato in modo che lo faccia alla membrana plasmatica. Quindi, ivacaftor gli dà il “calcio” di cui ha bisogno per funzionare.
“Il delta F508 CFTR è ancora handicappato, ma è meglio di niente”, afferma Guggino. “E questo significa che non stiamo solo trattando i sintomi più. Stiamo trattando le cause alla radice.”
Nessun bambino lasciato indietro
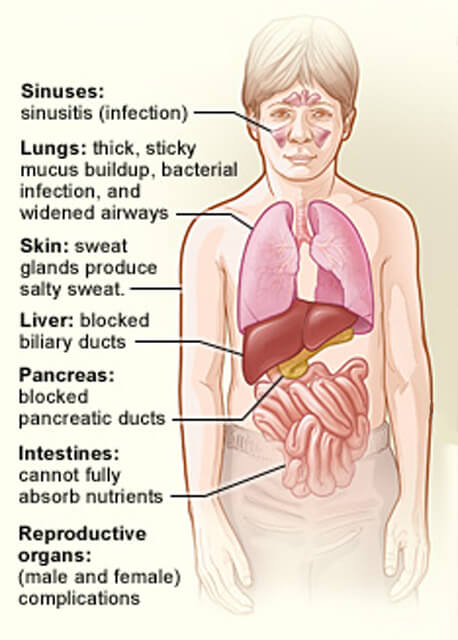 Gli organi colpiti dalla fibrosi cistica.
Gli organi colpiti dalla fibrosi cistica.Credito: National Heart, Lung and Blood Institute
Oltre il 40% dei pazienti con CF con altre mutazioni — alcune piuttosto rare — non sono sempre così fortunati. Ci sono oltre 1.700 mutazioni nel gene CFTR che causano la CF e solo una frazione minore è stata testata per la risposta ai farmaci approvati. Alcuni di quelli testati rispondono a uno o l’altro farmaco, ma altri no. Ancora, i ricercatori sono fiduciosi che un giorno saranno in grado di aiutare tutti. Taglio prevede infine categorizzare ogni paziente dal tipo, una parola che ha coniato per descrivere gruppi di pazienti che sono suscettibili di rispondere alla stessa terapia a causa di una causa sottostante comune dei loro sintomi. Ha lavorato a stretto contatto con i colleghi Patrick Sosnay e Karen Raraigh per estrarre dati da 88.000 individui in tutto il mondo per testare il suo concetto.
Possiamo utilizzare le informazioni generate da esperimenti nelle cellule per raggruppare le mutazioni che influenzano la stessa proprietà di CFTR e quindi dovrebbero rispondere allo stesso pannello di farmaci CFTR. Raggruppare le mutazioni in base al tipo consentirebbe studi clinici su pazienti portatori di mutazioni diverse, invece di studi clinici che valutano una mutazione alla volta”, afferma Cutting. “Questa è la medicina di precisione che diventa una realtà.”
Sfortunatamente, le terapie a base di farmaci non funzioneranno per tutti i pazienti con CF, specialmente la percentuale di 2 che non produce affatto CFTR. Per questi, i ricercatori stanno sviluppando modi per indirizzare il gene mutato stesso, anche se ci sono ancora molti ostacoli da superare.
Guggino ha elaborato un sistema di terapia genica che utilizza un virus adeno-associato modificato (AAV) per depositare una buona versione del gene CFTR all’interno delle cellule. Il sistema si è dimostrato nelle cellule delle vie aeree umane e nei roditori.
Provando un approccio diverso, Liudmila Cebotaru, del Dipartimento di Medicina, ha ideato un nuovo modo per combinare la terapia genica e la riparazione delle proteine con un meccanismo chiamato transcomplementazione. Invece di posizionare il gene CFTR full-length all’interno di AAV, lei sta usando una versione più corta che è più facilmente inserito nel genoma della cellula. Quando viene prodotta la proteina più corta, si lega alla proteina mutante del paziente e la aiuta a raggiungere la membrana plasmatica. Sia Cebotaru che Guggino stanno ora testando il suo nuovo approccio nelle scimmie rhesus perché i loro polmoni e il loro sistema immunitario sono molto vicini agli umani. Sperano di iniziare una sperimentazione clinica nei prossimi anni, se tutto va bene.
” Mi piace pensare che salti la batteria della tua auto”, dice. “Con un piccolo aiuto in più, le proteine CFTR dei pazienti possono arrivare a destinazione.”
Per i ricercatori e i pazienti, la destinazione non è altro che una cura per la CF. E anche se è ancora molto lontano, ci sono segnali incoraggianti che possiamo arrivarci.