door Catherine Gara
januari 2016—slijm is misschien niet iets waar we graag over nadenken, maar ons leven hangt ervan af. Bij cystic fibrosis (CF), de verdikking van slijm leidt tot longinfecties en darmblokken, onder andere symptomen. De ziekte wordt veroorzaakt door één enkel gen dat wordt beïnvloed door één of meer van de meer dan 1.700 mutaties waarvan bekend is dat ze de aandoening veroorzaken. Bij Johns Hopkins werken onderzoekers en clinician genetici samen om alles te leren wat ze kunnen over het gen, zijn eiwit en wat er mis gaat bij patiënten, wetende dat elk stukje informatie dat ze verzamelen hen dichter brengt bij het hebben van behandelingsopties voor alle individuen met CF.
geef me het zout
deze dagen zijn patiënten met CF veel beter af dankzij behandelingen die hun frequente longinfecties onder controle houden, maar een diagnose van CF betekent nog steeds een levensverwachting van slechts 38 vanwege de tol voor de alvleesklier, lever en darmen. Het probleem komt van defecten in het cystic fibrosis transmembrane conductance regulator (CFTR) gen, dat de blauwdruk voor het CFTR-eiwit draagt. Het CFTR-eiwit is als de brievenbus in een voordeur. Het vormt een kleine doorgang tussen de binnen-en buitenkant van de cel.
wanneer het goed werkt, helpt CFTR de passage van chloride-ionen (een bestanddeel van zout) in en uit de cel te controleren. In de longen en de kanalen van de alvleesklier, wanneer chloride cellen verlaat, stimuleert het water om te volgen. Dat water helpt dunne lagen slijm te vormen. In de longen vangt het slijm stof en bacteriën op die er niet zouden moeten zijn. De trilharen, of haarachtige structuren, op de cellen die langs de longen lopen, leiden dan het slijm door de luchtweg naar de mond, waar het wordt ingeslikt en naar verteerd. In de alvleesklier helpt de vloeistof enzymen naar de darm te dragen om te helpen bij de spijsvertering. Als het slijm te stroperig is in de longen, kunnen de trilharen het er niet uit bewegen, zodat bacteriën er blijven om infecties te veroorzaken; als het te stroperig is in de alvleesklierkanalen, bereiken enzymen de darm niet en verteert voedsel niet goed.Garry Cutting, professor aan het Institute of Genetic Medicine, en Bill Guggino, directeur van de afdeling Fysiologie, hebben het CFTR-gen en het gecodeerde eiwit gedurende het grootste deel van hun loopbaan bestudeerd. Cutting ‘ s interesse komt voort uit de zorg voor een paar broers met CF terwijl een inwoner van Johns Hopkins. Guggino ‘ s interesse gaat nog verder terug—naar zijn jeugdreizen naar de zee. Hij vroeg zich af hoe vissen konden overleven in zout water, en hij leerde dat het korte antwoord is: hun versie van CFTR.
als het moeilijk lijkt om een hele carrière te focussen op een enkel gen en zijn eiwitproduct, zie het dan in plaats daarvan als een complexe Rubik ‘ s kubus gemaakt van een keten van 1.480 magnetische blokken (aminozuren). Een mutatie in het CFTR-gen betekent vaak een verandering in één van de aminozuren, die de uiteindelijke driedimensionale vorm dramatisch kan beïnvloeden.
sommige mutaties zorgen ervoor dat het eiwit niet wordt aangemaakt. Anderen staan een gedeeltelijke proteã ne toe om te worden samengesteld. Hoe dichter bij het begin van het eiwit deze mutaties optreden, hoe slechter voor zijn functie. Andere mutaties treden op precies de verkeerde plaats op en voorkomen bijvoorbeeld dat het zoutkanaal zich opent. Weer anderen maken CFTR vouwen onjuist, die signalen van de cel kwaliteitscontrole team om het op te halen en te recyclen. En anderen interfereren niet met de functie van het eiwit, maar ze voorkomen dat het naar het plasmamembraan komt waar het zijn werk doet.
Cutting en guggino ‘ s inspanningen hebben bijgedragen tot het ontwerp van twee CF-geneesmiddelen op de markt: ivacaftor en lumacaftor. Ivacaftor activeert CFTR met de mutatie G551D. Het laboratorium van Cutting rapporteerde deze mutatie voor het eerst in 1990, en Guggino en Cutting genereerden vervolgens nieuwe inzichten in het effect van deze mutatie op de CFTR-functie en de symptomen van patiënten. Helaas wordt de G551D mutatie gevonden bij slechts 4 procent van de patiënten met CF. Het blijkt echter dat 50 procent van de patiënten met CF twee kopieën hebben van een andere mutatie (delta F508 genaamd), waardoor CFTR slecht wordt gevormd en naar de recyclagebak van de cel wordt gestuurd. Lumacaftor voorkomt dat het wordt gerecycled zodat het naar het plasmamembraan komt. Dan geeft ivacaftor het de “kick” die het nodig heeft om te werken.
“De delta F508 CFTR is nog steeds gehandicapt, maar het is beter dan niets,” zegt Guggino. “En dit betekent dat we niet alleen de behandeling van symptomen meer. We behandelen de onderliggende oorzaken.”
geen kind achtergelaten
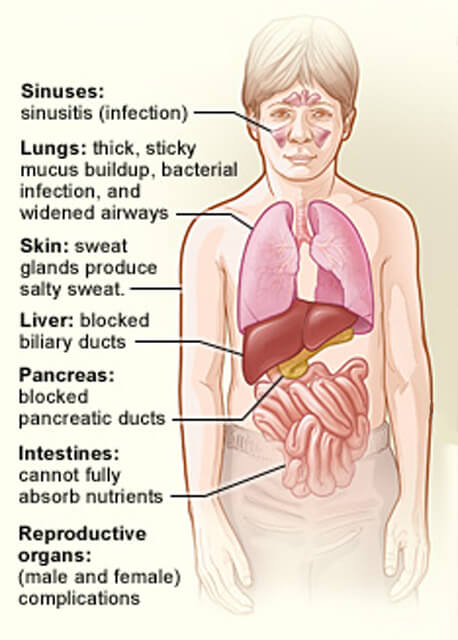 de organen aangetast door cystische fibrose.
de organen aangetast door cystische fibrose.krediet: National Heart, Lung and Blood Institute
meer dan 40 procent van de patiënten met CF met andere mutaties — sommige vrij zeldzaam — hebben niet altijd even veel geluk. Er zijn meer dan 1.700 mutaties in het CFTR gen die CF veroorzaken, en slechts een kleine fractie is getest op respons op de goedgekeurde geneesmiddelen. Sommige van die geteste reageren op de ene of de andere drug, maar anderen niet. toch zijn de onderzoekers hoopvol dat ze op een dag in staat zullen zijn om iedereen te helpen. Het snijden voorziet uiteindelijk het categoriseren van elke patiënt door theratype, een woord dat hij bedacht om groepen patiënten te beschrijven die waarschijnlijk op dezelfde therapie wegens een gemeenschappelijke onderliggende oorzaak van hun symptomen zullen reageren. Hij werkt nauw samen met collega ‘ s Patrick Sosnay en Karen Raraigh om gegevens van 88.000 individuen wereldwijd te delven om zijn concept te testen.
We kunnen informatie gegenereerd door experimenten in cellen gebruiken voor groepsmutaties die dezelfde eigenschap van CFTR beïnvloeden en daarom moeten reageren op hetzelfde panel van CFTR-geneesmiddelen. Het groeperen van mutaties volgens theratype zou klinische studies mogelijk maken bij patiënten met verschillende mutaties, in plaats van klinische studies die één mutatie per keer evalueren,” zegt Cutting. “Dat is precisiegeneeskunde die werkelijkheid wordt.”
helaas zullen geneesmiddelen gebaseerde therapieën niet werken voor alle patiënten met CF, vooral de 2 procent die helemaal geen CFTR maken. Voor deze, ontwikkelen de onderzoekers manieren om het gemuteerde gen zelf te richten, hoewel er nog vele hindernissen zijn te overwinnen.
Guggino heeft een gentherapiesysteem uitgewerkt dat een gemodificeerd adeno-geassocieerd virus (AAV) gebruikt om een goede versie van het CFTR-gen in cellen af te zetten. Het systeem heeft zich bewezen in menselijke luchtwegcellen en knaagdieren.Liudmila Cebotaru, van het Departement van Geneeskunde, probeerde een andere aanpak, en bedacht een nieuwe manier om gentherapie en eiwitreparatie te combineren met een mechanisme dat transcomplementatie wordt genoemd. In plaats van het volledige CFTR-gen binnen AAV te plaatsen, gebruikt ze een kortere versie die gemakkelijker in het genoom van de cel wordt ingebracht. Wanneer de kortere proteã ne wordt geproduceerd, bindt het aan de mutantproteã ne van de patiënt en helpt het bij het plasmamembraan. Zowel Cebotaru als Guggino testen nu haar nieuwe aanpak bij resusapen omdat hun longen en immuunsysteem zeer dicht bij die van de mens staan. Ze hopen in de komende jaren een klinische proef te starten, als alles goed gaat.
“Ik zie het graag als het springen van de batterij van je auto,” zegt ze. “Met een beetje extra hulp kunnen de CFTR-eiwitten van de patiënten hun bestemming bereiken.”
voor zowel onderzoekers als patiënten is de bestemming niets minder dan een remedie voor CF. En hoewel het nog ver weg is, zijn er bemoedigende tekenen dat we er misschien komen.