av Catherine Gara
januari 2016-slem kanske inte är något vi tycker om att tänka på, men våra liv beror på det. Vid cystisk fibros (CF) leder förtjockningen av slem till lunginfektioner och tarmblock, bland andra symtom. Sjukdomen orsakas av en enda gen som påverkas av en eller flera av de mer än 1700 mutationer som är kända för att orsaka störningen. På Johns Hopkins, forskare och kliniker genetiker arbetar tillsammans för att lära sig allt de kan om genen, dess protein och vad som går fel hos patienter, att veta att varje information de samlar in för dem närmare att ha behandlingsalternativ för alla individer med CF.
Ge mig saltet
dessa dagar är patienter med CF mycket bättre tack vare behandlingar som hanterar deras frekventa lunginfektioner, men en diagnos av CF betyder fortfarande en livslängd på endast 38 på grund av den vägtull det tar på bukspottkörteln, lever och tarmar. Problemet kommer från defekter i cystisk fibros transmembran konduktansregulator (CFTR) gen, som bär ritningen för CFTR-proteinet. CFTR-proteinet är som brevinkastet i en ytterdörr. Det bildar en liten passage mellan insidan och utsidan av cellen.
När CFTR fungerar korrekt hjälper det till att kontrollera passagen av kloridjoner (en komponent av salt) in och ut ur cellen. I lungorna och kanalerna i bukspottkörteln, när klorid lämnar celler, uppmuntrar det vatten att följa. Att vatten hjälper till att bilda tunna lager av slem. I lungorna fångar slem damm och bakterier som inte borde vara där. Cilia, eller hårliknande strukturer, på cellerna som leder lungorna, herdar sedan slem upp i luftvägarna till munnen, där det sväljs och skickas för att smälta. I bukspottkörteln hjälper vätskan att bära enzymer till tarmen för att hjälpa till med matsmältningen. Om slemet är för visköst i lungorna kan cilia inte flytta ut det, så bakterier förblir där för att orsaka infektioner; om det är för visköst i bukspottkörtelkanalerna når enzymer inte tarmen och maten smälter inte ordentligt.
a Protein ’ s Path
Garry Cutting, professor vid Institutet för genetisk medicin, och Bill Guggino, chef för Institutionen för fysiologi, har studerat CFTR-genen och dess kodade protein under de flesta av sina karriärer. Cutting intresse härrör från att ta hand om ett par bröder med CF medan bosatt på Johns Hopkins. Gugginos intresse går tillbaka ännu längre-till hans pojkresor till havet. Han undrade hur fisk kunde överleva i saltvatten, och han lärde sig att det korta svaret är: deras version av CFTR.
om det verkar svårt att föreställa sig att fokusera en hel karriär på en enda gen och dess proteinprodukt, tänk på det istället som en komplex Rubiks kub Tillverkad av en kedja av 1 480 magnetiska block (aminosyror). En mutation i CFTR-genen betyder ofta en förändring i en av aminosyrorna, vilket dramatiskt kan påverka den slutliga tredimensionella formen.
vissa mutationer gör att proteinet inte görs. Andra tillåter att ett partiellt protein syntetiseras. Ju närmare proteinets början dessa mutationer inträffar, desto sämre för dess funktion. Andra mutationer uppträder på fel plats och förhindrar till exempel att saltkanalen öppnas. Ytterligare andra gör att CFTR viks felaktigt, vilket signalerar cellens kvalitetskontrollteam för att hämta det och återvinna det. Och andra stör inte proteinets funktion, men de hindrar det från att komma till plasmamembranet där det gör sitt arbete.
Cutting och Gugginos ansträngningar har bidragit till utformningen av två CF-läkemedel på marknaden: ivacaftor och lumacaftor. Ivacaftor aktiverar CFTR med mutationen G551D. Cutting ’ s lab rapporterade först denna mutation 1990, och Guggino och Cutting genererade därefter nya insikter om effekten av denna mutation på CFTR-funktion och patienters symtom. Tyvärr finns G551D-mutationen hos endast 4 procent av patienterna med CF. Det visar sig dock att 50 procent av patienterna med CF har två kopior av en annan mutation (kallad delta F508), vilket gör att CFTR bildas dåligt och skickas till cellens papperskorgen. Lumacaftor förhindrar att det återvinns så att det kommer till plasmamembranet. Sedan ger ivacaftor den” kick ” den behöver för att fungera.
”delta F508 CFTR är fortfarande handikappad, men det är bättre än ingenting”, säger Guggino. ”Och det betyder att vi inte bara behandlar symtom längre. Vi behandlar grundorsakerna.”
inget barn kvar
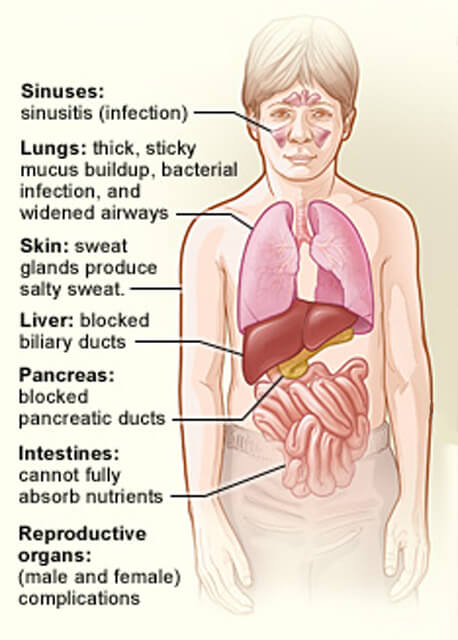 de organ som påverkas av cystisk fibros.
de organ som påverkas av cystisk fibros.kredit: National Heart, Lung and Blood Institute
de mer än 40 procent av patienterna med CF med andra mutationer — några ganska sällsynta — är inte alltid lika lyckliga. Det finns över 1700 mutationer i CFTR-genen som orsakar CF, och endast en mindre fraktion har testats för svar på de godkända läkemedlen. Några av de testade svarar på det ena eller det andra läkemedlet, men andra gör det inte. ändå är forskarna hoppfulla att de en dag kommer att kunna hjälpa alla. Cutting förutser så småningom att kategorisera varje patient efter theratype, ett ord som han myntade för att beskriva grupper av patienter som sannolikt kommer att svara på samma terapi på grund av en vanlig bakomliggande orsak till deras symtom. Han har arbetat nära med kollegorna Patrick Sosnay och Karen Raraigh för att mina data från 88 000 individer över hela världen för att testa sitt koncept.
Vi kan använda information som genereras av experiment i celler för att gruppera mutationer som påverkar samma egenskap hos CFTR och därför bör svara på samma panel av CFTR-läkemedel. Gruppering av mutationer enligt theratype skulle möjliggöra kliniska prövningar på patienter som bär olika mutationer, istället för kliniska prövningar som utvärderar en mutation i taget”, säger Cutting. ”Det är precisionsmedicin som blir verklighet.”
tyvärr kommer läkemedelsbaserade terapier inte att fungera för alla patienter med CF, särskilt de 2 procent som inte gör någon CFTR alls. För dessa utvecklar forskare sätt att rikta sig mot den muterade genen själv, men det finns fortfarande många hinder att övervinna.
Guggino har utarbetat ett genterapisystem som använder ett modifierat adenoassocierat virus (AAV) för att deponera en bra version av CFTR-genen inuti celler. Systemet har visat sig i mänskliga luftvägsceller och gnagare.
Liudmila Cebotaru, från Institutionen för medicin, försökte ett annat tillvägagångssätt, utarbetade ett nytt sätt att kombinera genterapi och proteinreparation med en mekanism som kallas transkomplementering. Istället för att placera CFTR-genen i full längd i AAV använder hon en kortare version som lättare sätts in i cellens genom. När det kortare proteinet produceras binder det till patientens mutanta protein och hjälper det att komma till plasmamembranet. Både Cebotaru och Guggino testar nu sitt nya tillvägagångssätt i rhesusapor eftersom deras lungor och immunsystem är mycket nära människor. De hoppas kunna starta en klinisk prövning de närmaste åren, om allt går bra.
”Jag gillar att tänka på det som att hoppa på bilens batteri”, säger hon. ”Med lite extra hjälp kan patienternas CFTR-proteiner komma till sin destination.”
för både forskare och patienter är destinationen inget mindre än ett botemedel mot CF. Och även om det fortfarande är långt borta, det finns uppmuntrande tecken på att vi kan komma dit.