Von Catherine Gara
Januar 2016 — Schleim ist vielleicht nicht etwas, worüber wir gerne nachdenken, aber unser Leben hängt davon ab. Bei Mukoviszidose (CF) führt die Verdickung des Schleims unter anderem zu Lungeninfektionen und Darmblockaden. Die Krankheit wird durch ein einzelnes Gen verursacht, das von einer oder mehreren der mehr als 1.700 Mutationen betroffen ist, von denen bekannt ist, dass sie die Störung verursachen. An der Johns Hopkins, Forscher und Klinikgenetiker arbeiten zusammen, um alles über das Gen zu erfahren, sein Protein und was bei Patienten schief geht, Zu wissen, dass jede Information, die sie sammeln, sie näher an Behandlungsmöglichkeiten für alle Menschen mit CF bringt.
Gib mir das Salz
Heutzutage sind Patienten mit CF dank Behandlungen, die ihre häufigen Lungeninfektionen behandeln, viel besser dran, aber eine Diagnose von CF bedeutet immer noch eine Lebenserwartung von nur 38 aufgrund der Belastung von Bauchspeicheldrüse, Leber und Darm. Das Problem kommt von Defekten im CFTR-Gen (Cystic Fibrosis Transmembrane Conductance Regulator), das den Bauplan für das CFTR-Protein trägt. Das CFTR-Protein ist wie der Postschlitz in einer Haustür. Es bildet einen kleinen Durchgang zwischen dem Inneren und dem Äußeren der Zelle.
Bei ordnungsgemäßer Funktion hilft die CFTR, den Durchgang von Chloridionen (einer Salzkomponente) in und aus der Zelle zu kontrollieren. Wenn Chlorid in der Lunge und in den Pankreasgängen die Zellen verlässt, fördert es das Folgen von Wasser. Dieses Wasser hilft, dünne Schleimschichten zu bilden. In der Lunge fängt der Schleim Staub und Bakterien ein, die nicht da sein sollten. Die Zilien oder haarähnlichen Strukturen auf den Zellen, die die Lunge auskleiden, leiten den Schleim dann durch die Atemwege zum Mund, wo er verschluckt und zur Verdauung geschickt wird. In der Bauchspeicheldrüse trägt die Flüssigkeit dazu bei, Enzyme in den Darm zu transportieren, um die Verdauung der Nahrung zu unterstützen. Wenn der Schleim in der Lunge zu viskos ist, können die Zilien ihn nicht herausbewegen, sodass Bakterien dort verbleiben, um Infektionen zu verursachen.
Der Weg eines Proteins
Garry Cutting, Professor am Institut für Genetische Medizin, und Bill Guggino, Direktor der Abteilung für Physiologie, haben das CFTR-Gen und sein kodiertes Protein die meiste Zeit ihrer Karriere untersucht. Cuttings Interesse rührt von der Pflege eines Bruderpaares mit CF während eines Aufenthalts in Johns Hopkins her. Gugginos Interesse geht noch weiter zurück – zu seinen Jugendausflügen ans Meer. Er fragte sich, wie Fische in Salzwasser überleben könnten, und er erfuhr, dass die kurze Antwort lautet: ihre Version von CFTR.
Wenn es schwer vorstellbar ist, eine ganze Karriere auf ein einzelnes Gen und sein Proteinprodukt zu konzentrieren, stellen Sie sich stattdessen einen komplexen Zauberwürfel vor, der aus einer Kette von 1.480 Magnetblöcken (Aminosäuren) besteht. Eine Mutation im CFTR-Gen bedeutet oft eine Veränderung einer der Aminosäuren, die die endgültige dreidimensionale Form dramatisch beeinflussen kann.
Einige Mutationen führen dazu, dass das Protein nicht hergestellt wird. Andere erlauben die Synthese eines partiellen Proteins. Je näher am Anfang des Proteins diese Mutationen auftreten, desto schlechter für seine Funktion. Andere Mutationen treten genau an der falschen Stelle auf und verhindern beispielsweise, dass sich der Salzkanal öffnet. Wieder andere lassen CFTR falsch falten, was dem Qualitätskontrollteam der Zelle signalisiert, es aufzunehmen und zu recyceln. Und andere stören die Funktion des Proteins nicht, aber sie verhindern, dass es zur Plasmamembran gelangt, wo es seine Arbeit verrichtet.
Die Bemühungen von Cutting und Guggino haben zur Entwicklung von zwei CF-Medikamenten auf dem Markt beigetragen: Ivacaftor und Lumacaftor. Ivacaftor aktiviert CFTR mit der Mutation G551D. Cuttings Labor berichtete erstmals 1990 über diese Mutation, und Guggino und Cutting generierten anschließend neue Erkenntnisse über die Wirkung dieser Mutation auf die CFTR-Funktion und die Symptome der Patienten. Leider wird die G551D-Mutation nur bei 4 Prozent der Patienten mit CF gefunden. Es stellt sich jedoch heraus, dass 50 Prozent der Patienten mit CF zwei Kopien einer anderen Mutation (Delta F508 genannt) haben, die dazu führt, dass CFTR schlecht gebildet und in den Recyclingbehälter der Zelle geschickt wird. Lumacaftor verhindert, dass es recycelt wird, so dass es zur Plasmamembran gelangt. Dann gibt Ivacaftor ihm den „Kick“, den es braucht, um zu funktionieren.
„Der Delta F508 CFTR ist immer noch behindert, aber besser als nichts“, sagt Guggino. „Und das bedeutet, dass wir nicht mehr nur Symptome behandeln. Wir behandeln die Ursachen.“
Kein Kind zurückgelassen
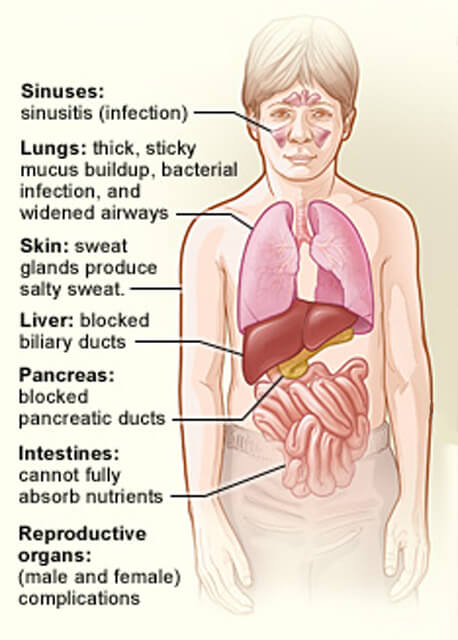 Die von Mukoviszidose betroffenen Organe.
Die von Mukoviszidose betroffenen Organe.Credit: National Heart, Lung and Blood Institute
Die mehr als 40 Prozent der Patienten mit CF mit anderen Mutationen — einige recht selten — sind nicht immer so viel Glück. Es gibt über 1.700 Mutationen im CFTR-Gen, die CF verursachen, und nur ein geringer Teil wurde auf das Ansprechen auf die zugelassenen Medikamente getestet. Einige der Getesteten sprechen auf das eine oder andere Medikament an, andere nicht. Dennoch hoffen die Forscher, dass sie eines Tages allen helfen können. Cutting sieht vor, jeden Patienten schließlich nach Theratyp zu kategorisieren, ein Wort, das er geprägt hat, um Gruppen von Patienten zu beschreiben, die aufgrund einer gemeinsamen Ursache ihrer Symptome wahrscheinlich auf dieselbe Therapie ansprechen. Er hat eng mit seinen Kollegen Patrick Sosnay und Karen Raraigh zusammengearbeitet, um Daten von 88.000 Personen weltweit zu sammeln und sein Konzept zu testen.Wir können Informationen verwenden, die durch Experimente in Zellen generiert wurden, um Mutationen zu gruppieren, die dieselbe Eigenschaft von CFTR beeinflussen und daher auf dieselbe Gruppe von CFTR-Medikamenten ansprechen sollten. Die Gruppierung von Mutationen nach Theratyp würde klinische Studien an Patienten mit unterschiedlichen Mutationen ermöglichen, anstatt klinische Studien, die jeweils eine Mutation bewerten „, sagt Cutting. „Präzisionsmedizin wird Realität.“Leider werden medikamentöse Therapien nicht für alle Patienten mit CF funktionieren, besonders nicht für die 2 Prozent, die überhaupt keine CFTR machen. Für diese entwickeln Forscher Möglichkeiten, das mutierte Gen selbst anzugreifen, obwohl noch viele Hürden zu überwinden sind.
Guggino hat ein Gentherapiesystem entwickelt, das ein modifiziertes Adeno-assoziiertes Virus (AAV) verwendet, um eine gute Version des CFTR-Gens in Zellen abzulegen. Das System hat sich in menschlichen Atemwegszellen und Nagetieren bewährt.Liudmila Cebotaru vom Department of Medicine versuchte einen anderen Ansatz und entwickelte einen neuartigen Weg, Gentherapie und Proteinreparatur durch einen Mechanismus namens Transcomplementation zu kombinieren. Anstatt das CFTR-Gen in voller Länge innerhalb einer Zelle zu platzieren, verwendet sie eine kürzere Version, die leichter in das Genom der Zelle eingefügt werden kann. Wenn das kürzere Protein produziert wird, bindet es an das mutierte Protein des Patienten und hilft ihm, an die Plasmamembran zu gelangen. Sowohl Cebotaru als auch Guggino testen jetzt ihren neuen Ansatz bei Rhesusaffen, da ihre Lungen und ihr Immunsystem dem Menschen sehr nahe kommen. Sie hoffen, in den nächsten Jahren mit einer klinischen Studie beginnen zu können, wenn alles gut geht.
„Ich stelle es mir gerne so vor, als würde ich die Batterie deines Autos springen lassen“, sagt sie. „Mit ein wenig zusätzlicher Hilfe können die CFTR-Proteine der Patienten an ihr Ziel gelangen.“Für die Forscher und Patienten gleichermaßen ist das Ziel nichts weniger als eine Heilung für CF. Und obwohl es noch ein weiter Weg ist, gibt es ermutigende Anzeichen, dass wir dorthin gelangen können.