Catherine Gara
tammikuu 2016—limaa ei ehkä haluta ajatella, mutta elämämme riippuu siitä. Kystisessä fibroosissa (CF) liman paksuuntuminen johtaa muun muassa keuhkotulehduksiin ja suolilohkoihin. Taudin aiheuttaa yksittäinen geeni, johon vaikuttaa yksi tai useampi niistä yli 1 700 mutaatiosta, joiden tiedetään aiheuttavan häiriötä. Johns Hopkinsissa tutkijat ja kliinikkogeneetikot työskentelevät yhdessä oppiakseen kaiken mahdollisen geenistä, sen proteiinista ja siitä, mikä menee pieleen potilailla, tietäen, että jokainen heidän keräämänsä tieto tuo heidät lähemmäksi hoitovaihtoehtoja kaikille CF-henkilöille.
Anna minulle suola
nykyään CF: ää sairastavat potilaat voivat paljon paremmin toistuvien keuhkotulehdustensa hoitojen ansiosta, mutta CF: n diagnoosi merkitsee silti elinajanodotetta, joka on vain 38, koska se verottaa haimaa, maksaa ja suolistoa. Ongelmia tulee vikoja kystinen fibroosi transmembrane conductance regulator (CFTR) geeni, joka kuljettaa suunnitelma CFTR proteiinia. CFTR-proteiini on kuin etuoven postiluukku. Se muodostaa pienen käytävän solun sisä-ja ulkopuolelle.
kun CFTR toimii oikein, se auttaa hallitsemaan kloridi-ionien (suolan komponentti) kulkua soluun ja ulos. Kun kloridi lähtee soluista keuhkoissa ja haiman tiehyissä, se kannustaa vettä seuraamaan. Vesi auttaa muodostamaan ohuita limakerroksia. Keuhkoihin Lima vangitsee pölyä ja bakteereja, joita siellä ei pitäisi olla. Keuhkoja reunustavien solujen värekarvat eli karvamaiset rakenteet paimentavat sitten limaa hengitysteitä pitkin suuhun, missä se niellään ja lähetetään sulatettavaksi. Haimassa neste auttaa kuljettamaan entsyymejä suolistoon auttamaan ruoansulatuksessa. Jos lima on liian viskoosia keuhkoissa, värekarvat eivät voi siirtää sitä ulos, joten bakteerit jäävät sinne aiheuttamaan infektioita; jos se on liian viskoosia haiman tiehyissä, entsyymit eivät pääse suoleen, ja ruoka ei sula kunnolla.
a Protein ’ s Path
genetiikan instituutin professori Garry Cutting ja fysiologian laitoksen johtaja Bill Guggino ovat tutkineet CFTR-geeniä ja sen koodattua proteiinia suurimman osan urastaan. Cuttingin kiinnostus johtuu siitä, että hän hoitaa VELJESPARIA CF: n kanssa asuessaan Johns Hopkinsissa. Gugginon kiinnostus ulottuu vielä kauemmas-hänen poikavuosiensa matkoille merelle. Hän ihmetteli, miten kalat voisivat selviytyä suolavedessä, ja sai tietää, että lyhyt vastaus on: heidän versionsa CFTR: stä.
Jos tuntuu vaikealta kuvitella keskittyvänsä koko uran yhteen geeniin ja sen proteiinituotteeseen, ajattele sitä sen sijaan monimutkaisena Rubikin kuutiona, joka on tehty 1 480 magneettisen lohkon (aminohapon) ketjusta. CFTR-geenin mutaatio tarkoittaa usein jonkin aminohapon muutosta, joka voi dramaattisesti vaikuttaa lopulliseen kolmiulotteiseen muotoon.
jotkin mutaatiot aiheuttavat sen, että proteiinia ei synny. Toiset mahdollistavat osittaisen proteiinin syntetisoinnin. Mitä lähempänä alussa proteiinin nämä mutaatiot tapahtuvat, sitä huonompi sen toiminta. Muut mutaatiot tapahtuvat juuri väärässä paikassa ja estävät esimerkiksi suolakanavan avautumisen. Toiset taas saavat CFTR: n taittumaan väärin, mikä viestii solun laadunvalvontatiimin poimivan sen talteen ja kierrättävän sen. Toiset taas eivät häiritse proteiinin toimintaa, mutta estävät sitä pääsemästä plasmakalvoon, jossa se tekee työnsä.
Cuttingin ja Gugginon ponnistelut ovat vaikuttaneet siihen, että markkinoille on suunniteltu kaksi CF-lääkettä: ivacaftor ja lumacaftor. Ivacaftor aktivoi CFTR: n, jolla on g551d-mutaatio. Cuttingin laboratorio raportoi tästä mutaatiosta ensimmäisen kerran vuonna 1990, ja Guggino ja Cutting loivat myöhemmin uusia näkemyksiä tämän mutaation vaikutuksesta CFTR: n toimintaan ja potilaiden oireisiin. Valitettavasti g551d-mutaatio löytyy vain 4 prosentilta CF-potilaista. On kuitenkin käynyt ilmi, että 50 prosentilla CF-potilaista on kaksi kopiota eri mutaatiosta (nimeltään delta F508), jonka vuoksi CFTR: ää muodostuu huonosti ja se lähetetään solun kierrätysastiaan. Lumacaftor estää sitä kierrätetään niin, että se tekee sen plasmakalvo. Sitten ivacaftor antaa sille ”potkua”, jota se tarvitsee toimiakseen.
”Delta F508 CFTR on edelleen invalidi, mutta parempi kuin ei mitään”, guggino sanoo. ”Ja tämä tarkoittaa, että emme enää vain hoida oireita. Hoidamme juurisyitä.”
lasta ei jäänyt
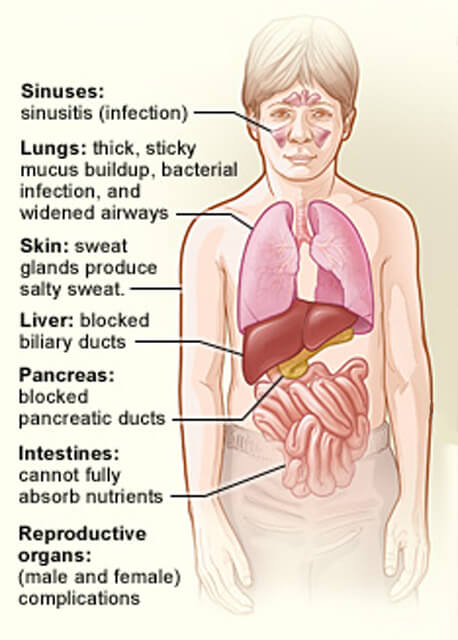 kystisen fibroosin vaikutuspiirissä olevat elimet.
kystisen fibroosin vaikutuspiirissä olevat elimet.luotto: National Heart, Lung and Blood Institute
yli 40 prosenttia CF — potilaista, joilla on muita mutaatioita — jotkut melko harvinaisia-eivät ole aina yhtä onnekkaita. CFTR-geenissä on yli 1 700 mutaatiota, jotka aiheuttavat CF: ää, ja vain pieni osa on testattu vasteen saamiseksi hyväksytyille lääkkeille. Jotkut testatuista reagoivat jompaankumpaan lääkkeeseen, mutta toiset eivät. Cutting ennakoi, että lopulta jokainen potilas luokitellaan tyypin mukaan, sanan, jonka hän keksi kuvaamaan potilasryhmiä, jotka todennäköisesti reagoivat samaan hoitoon, koska heidän oireidensa taustalla on yhteinen syy. Hän on tehnyt tiivistä yhteistyötä kollegoidensa Patrick Sosnayn ja Karen Raraighin kanssa louhiakseen 88 000 ihmisen tietoja maailmanlaajuisesti testatakseen konseptiaan.
Voimme käyttää solukokeiden tuottamaa tietoa ryhmitelläksemme mutaatioita, jotka vaikuttavat samaan CFTR: n ominaisuuteen, ja siksi meidän pitäisi reagoida samaan CFTR-lääkkeiden paneeliin. Mutaatioiden ryhmittely theratyypin mukaan mahdollistaisi kliiniset tutkimukset potilailla, joilla on erilaisia mutaatioita, sen sijaan, että kliinisessä tutkimuksessa arvioidaan yksi mutaatio kerrallaan, Cutting sanoo. ”Se on täsmälääketiede tulossa todellisuutta.”
valitettavasti lääkkeisiin perustuvat hoidot eivät toimi kaikille CF-potilaille, erityisesti niille 2 prosentille, jotka eivät tee CFTR: ää lainkaan. Näitä varten tutkijat kehittävät tapoja kohdentaa itse mutatoitunut geeni, vaikka on vielä monia esteitä voitettavana.
Guggino on kehittänyt geeniterapiajärjestelmän, joka käyttää muunneltua adenoon liittyvää virusta (AAV) tallettaakseen hyvän version CFTR-geenistä solujen sisään. Järjestelmä on osoittautunut ihmisen hengitysteiden soluissa ja jyrsijöissä.
kokeillessaan erilaista lähestymistapaa Liudmila Cebotaru lääketieteen laitokselta keksi uudenlaisen tavan yhdistää geeniterapia ja proteiinin korjaus mekanismin avulla, jota kutsutaan transkomplementaatioksi. Sen sijaan, että hän sijoittaisi täyspitkän CFTR-geenin AAV: hen, hän käyttää lyhyempää versiota, joka työnnetään helpommin solun genomiin. Kun lyhyempi proteiini tuotetaan, se sitoutuu potilaan mutanttiproteiiniin ja auttaa sitä pääsemään plasmakalvoon. Sekä Cebotaru että Guggino testaavat nyt hänen uutta lähestymistapaansa reesusapinoilla, koska niiden keuhkot ja immuunijärjestelmä ovat hyvin lähellä ihmistä. He toivovat pääsevänsä kliiniseen kokeeseen lähivuosina, jos kaikki menee hyvin.
”tykkään ajatella, että se on auton akun hyppimistä”, hän sanoo. ”Pienellä lisäavulla potilaiden CFTR-proteiinit pääsevät perille.”
sekä tutkijoille että potilaille määränpäänä ei ole vähempää kuin CF: n hoito. Ja vaikka siihen on vielä pitkä matka, on rohkaisevia merkkejä siitä, että voimme päästä perille.