Par Catherine Gara
Janvier 2016 – Le mucus n’est peut-être pas quelque chose auquel nous aimons penser, mais nos vies en dépendent. Dans la fibrose kystique (FK), l’épaississement du mucus entraîne des infections pulmonaires et des blocages intestinaux, entre autres symptômes. La maladie est causée par un seul gène affecté par une ou plusieurs des plus de 1 700 mutations connues pour causer le trouble. À Johns Hopkins, les chercheurs et les généticiens cliniciens travaillent ensemble pour apprendre tout ce qu’ils peuvent sur le gène, sa protéine et ce qui ne va pas chez les patients, sachant que chaque information qu’ils recueillent les rapproche des options de traitement pour toutes les personnes atteintes de mucoviscidose.
Passez-moi le sel
De nos jours, les patients atteints de mucoviscidose sont beaucoup mieux lotis grâce à des traitements qui gèrent leurs infections pulmonaires fréquentes, mais un diagnostic de mucoviscidose signifie toujours une espérance de vie de seulement 38 ans en raison du péage qu’il prend sur le pancréas, le foie et les intestins. Le problème vient des défauts du gène régulateur de conductance transmembranaire de la fibrose kystique (CFTR), qui porte le modèle de la protéine CFTR. La protéine CFTR est comme la fente de courrier dans une porte d’entrée. Il forme un petit passage entre l’intérieur et l’extérieur de la cellule.
Lorsqu’il fonctionne correctement, le CFTR aide à contrôler le passage des ions chlorure (un composant du sel) dans et hors de la cellule. Dans les poumons et les canaux du pancréas, lorsque le chlorure quitte les cellules, il encourage l’eau à suivre. Cette eau aide à former de fines couches de mucus. Dans les poumons, le mucus emprisonne la poussière et les bactéries qui ne devraient pas être là. Les cils, ou structures ressemblant à des cheveux, sur les cellules qui tapissent les poumons, transportent ensuite le mucus dans les voies respiratoires jusqu’à la bouche, où il est avalé et envoyé pour être digéré. Dans le pancréas, le liquide aide à transporter les enzymes vers l’intestin pour faciliter la digestion des aliments. Si le mucus est trop visqueux dans les poumons, les cils ne peuvent pas le sortir, de sorte que les bactéries y restent pour provoquer des infections; s’il est trop visqueux dans les canaux pancréatiques, les enzymes n’atteignent pas l’intestin et les aliments ne digèrent pas correctement.
Le chemin d’une protéine
Garry Cutting, professeur à l’Institut de médecine génétique, et Bill Guggino, directeur du Département de physiologie, ont étudié le gène CFTR et sa protéine codée pendant la majeure partie de leur carrière. L’intérêt de Cutting provient de la prise en charge d’une paire de frères atteints de mucoviscidose alors qu’ils résidaient à Johns Hopkins. L’intérêt de Guggino remonte encore plus loin — à ses voyages d’enfance à la mer. Il s’est demandé comment les poissons pouvaient survivre dans l’eau salée, et il a appris que la réponse courte était: leur version du CFTR.
S’il semble difficile d’imaginer concentrer toute une carrière sur un seul gène et son produit protéique, considérez-le plutôt comme un Rubik’s cube complexe fabriqué à partir d’une chaîne de 1480 blocs magnétiques (acides aminés). Une mutation du gène CFTR signifie souvent un changement dans l’un des acides aminés, ce qui peut affecter considérablement la forme tridimensionnelle finale.
Certaines mutations empêchent la fabrication de la protéine. D’autres permettent de synthétiser une protéine partielle. Plus ces mutations se rapprochent du début de la protéine, plus sa fonction est mauvaise. D’autres mutations se produisent au mauvais endroit et empêchent le canal de sel de s’ouvrir, par exemple. D’autres encore font plier le CFTR de manière incorrecte, ce qui indique à l’équipe de contrôle de la qualité de la cellule de le ramasser et de le recycler. Et d’autres n’interfèrent pas avec la fonction de la protéine, mais l’empêchent d’atteindre la membrane plasmique où elle fait son travail.
Les efforts de Cutting et de Guggino ont contribué à la conception de deux médicaments contre la mucoviscidose sur le marché : l’ivacaftor et le lumacaftor. L’Ivacaftor active le CFTR porteur de la mutation G551D. Le laboratoire de Cutting a signalé cette mutation pour la première fois en 1990, et Guggino et Cutting ont ensuite généré de nouvelles informations sur l’effet de cette mutation sur la fonction CFTR et les symptômes des patients. Malheureusement, la mutation G551D n’est trouvée que chez 4% des patients atteints de mucoviscidose. Cependant, il s’avère que 50% des patients atteints de mucoviscidose ont deux copies d’une mutation différente (appelée delta F508), ce qui entraîne une formation médiocre du CFTR et son envoi dans le bac de recyclage de la cellule. Lumacaftor l’empêche d’être recyclé pour qu’il parvienne à la membrane plasmique. Ensuite, ivacaftor lui donne le ”coup de pied » dont il a besoin pour fonctionner.
» Le delta F508 CFTR est toujours handicapé, mais c’est mieux que rien », explique Guggino. « Et cela signifie que nous ne nous contentons plus de traiter les symptômes. Nous traitons les causes profondes.”
Aucun Enfant Laissé Pour compte
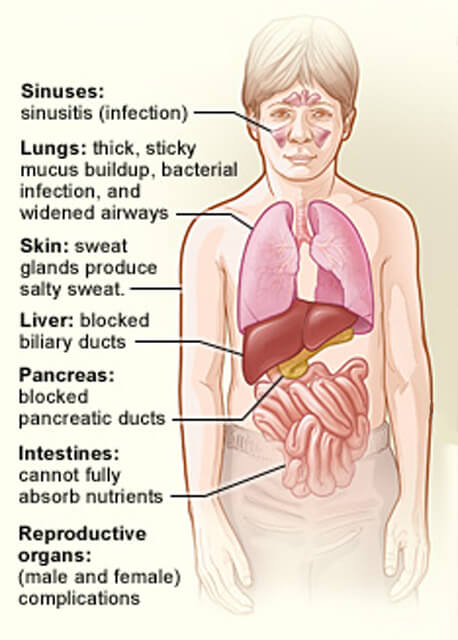 Les organes touchés par la mucoviscidose.
Les organes touchés par la mucoviscidose.Crédit: National Heart, Lung and Blood Institute
Plus de 40% des patients atteints de mucoviscidose avec d’autres mutations — certaines assez rares — n’ont pas toujours autant de chance. Il y a plus de 1 700 mutations dans le gène CFTR qui causent la FC, et seule une fraction mineure a été testée pour la réponse aux médicaments approuvés. Certains de ceux testés répondent à l’un ou l’autre médicament, mais d’autres non. Pourtant, les chercheurs espèrent qu’ils pourront un jour aider tout le monde. Cutting prévoit éventuellement de catégoriser chaque patient par type, un mot qu’il a inventé pour décrire des groupes de patients susceptibles de répondre à la même thérapie en raison d’une cause sous-jacente commune de leurs symptômes. Il a travaillé en étroite collaboration avec ses collègues Patrick Sosnay et Karen Raraigh pour extraire les données de 88 000 personnes dans le monde entier afin de tester son concept.
Nous pouvons utiliser les informations générées par des expériences dans des cellules pour regrouper des mutations qui affectent la même propriété du CFTR et devraient donc répondre au même panel de médicaments CFTR. Le regroupement des mutations en fonction du type de mutation permettrait des essais cliniques sur des patients porteurs de différentes mutations, au lieu d’essais cliniques qui évaluent une mutation à la fois ”, explique Cutting. » C’est la médecine de précision qui devient une réalité. »
Malheureusement, les thérapies médicamenteuses ne fonctionneront pas pour tous les patients atteints de mucoviscidose, en particulier les 2% qui ne font aucun CFTR. Pour ceux-ci, les chercheurs développent des moyens de cibler le gène muté lui-même, bien qu’il reste encore de nombreux obstacles à surmonter.
Guggino a mis au point un système de thérapie génique qui utilise un virus adéno-associé modifié (AAV) pour déposer une bonne version du gène CFTR à l’intérieur des cellules. Le système a fait ses preuves dans les cellules des voies respiratoires humaines et les rongeurs.
En essayant une approche différente, Liudmila Cebotaru, du Département de médecine, a conçu une nouvelle façon de combiner la thérapie génique et la réparation des protéines par un mécanisme appelé transcomplémentation. Au lieu de placer le gène CFTR sur toute la longueur dans AAV, elle utilise une version plus courte qui est plus facilement insérée dans le génome de la cellule. Lorsque la protéine la plus courte est produite, elle se lie à la protéine mutante du patient et l’aide à atteindre la membrane plasmique. Cebotaru et Guggino testent actuellement sa nouvelle approche chez les singes rhésus car leurs poumons et leur système immunitaire sont très proches de ceux des humains. Ils espèrent commencer un essai clinique dans les prochaines années, si tout se passe bien.
« J’aime penser que c’est sauter la batterie de votre voiture », dit-elle. « Avec un peu d’aide supplémentaire, les protéines CFTR des patients peuvent arriver à destination.”
Pour les chercheurs comme pour les patients, la destination n’est rien de moins qu’un remède contre la mucoviscidose. Et bien qu’il soit encore loin, il y a des signes encourageants que nous pourrions y arriver.