Av Catherine Gara
januar 2016—Slim kan ikke være noe vi liker å tenke på, men våre liv er avhengig av det. Ved cystisk fibrose (CF) fører fortykning av slim til lungeinfeksjoner og tarmblokker, blant andre symptomer. Sykdommen er forårsaket av et enkelt gen påvirket av en eller flere av de mer enn 1700 mutasjonene som er kjent for å forårsake uorden. På Johns Hopkins jobber forskere og kliniker genetikere sammen for å lære alt de kan om genet, dets protein og hva som går galt hos pasienter, og vet at hver informasjon de samler bringer dem nærmere å ha behandlingsmuligheter for alle personer med CF.I Disse dager er pasienter med CF mye bedre takket være behandlinger som håndterer deres hyppige lungeinfeksjoner, men en diagnose AV CF betyr fortsatt en forventet levetid på bare 38 på grunn av tollen det tar på bukspyttkjertelen, leveren og tarmene. Problemet kommer fra defekter i CYSTISK fibrose transmembrane konduktans regulator (CFTR) genet, som bærer blåkopi FOR CFTR protein. CFTR-proteinet er som postsporet i en inngangsdør. Den danner en liten passasje mellom innsiden og utsiden av cellen.NÅR CFTR fungerer som det skal, hjelper DET å kontrollere passasjen av kloridioner (en komponent av salt) inn og ut av cellen. I lungene og kanalene i bukspyttkjertelen, når klorid forlater celler, oppfordrer det vann til å følge. Det vannet bidrar til å danne tynne lag av slim. I lungene fanger slimet støv og bakterier som ikke burde være der. Flimmerhårene, eller hairlike strukturer, på cellene som linje lungene så gjeter slim opp luftveiene til munnen, hvor det blir svelget og sendt for å bli fordøyd. I bukspyttkjertelen hjelper væsken med å bære enzymer til tarmen for å hjelpe til med fordøyelsen. Hvis slimet er for viskøst i lungene, kan cilia ikke flytte det ut, så bakterier forblir der for å forårsake infeksjoner; hvis det er for viskøst i bukspyttkjertelen, kommer enzymer ikke til tarmen, og maten fordøyes ikke riktig.Garry Cutting, professor Ved Institutt For Genetisk Medisin, Og Bill Guggino, direktør Ved Institutt for Fysiologi, har studert CFTR-genet og dets kodede protein i de fleste av sine karrierer. Cutting interesse stammer fra omsorg for et par brødre MED CF mens bosatt På Johns Hopkins. Gugginos interesse går enda lenger tilbake-til hans barndomsturer til sjøen. Han lurte på hvordan fisk kunne overleve i saltvann, og han lærte at det korte svaret er: deres versjon AV CFTR.Hvis det virker vanskelig å forestille seg å fokusere en hel karriere på et enkelt gen og dets proteinprodukt, tenk på det i stedet som en kompleks Rubiks kube laget av en kjede av 1,480 magnetiske blokker (aminosyrer). En mutasjon I CFTR-genet vil ofte bety en endring i en av aminosyrene, noe som dramatisk kan påvirke den endelige tredimensjonale formen.
noen mutasjoner forårsaker at proteinet ikke blir laget. Andre tillater et delvis protein å bli syntetisert. Jo nærmere begynnelsen av proteinet disse mutasjonene oppstår, jo verre for sin funksjon. Andre mutasjoner forekommer på bare feil sted og forhindrer at saltkanalen åpnes, for eksempel. Atter andre gjør CFTR fold feil, som signaliserer cellens kvalitetskontroll team for å plukke den opp og resirkulere den. Og andre forstyrrer ikke proteinets funksjon, men de forhindrer det i å komme til plasmamembranen der den gjør sitt arbeid.Cutting Og Gugginos innsats har bidratt til utformingen av to cf-legemidler på markedet: ivacaftor og lumacaftor. Ivakaftor aktiverer CFTR med mutasjonen G551D. Cutting ‘ s lab rapporterte først denne mutasjonen i 1990, Og Guggino og Cutting genererte senere ny innsikt i effekten av denne mutasjonen på CFTR-funksjon og pasientens symptomer. Dessverre er g551d-mutasjonen funnet hos bare 4 prosent av pasientene MED CF. Det viser seg imidlertid at 50 prosent av pasientene med CF har to kopier av en annen mutasjon (kalt delta F508), noe som fører TIL AT CFTR blir dårlig dannet og sendt til cellens resirkuleringsboks. Lumacaftor hindrer det fra å bli resirkulert slik at det gjør det til plasmamembranen. Deretter gir ivacaftor det «kick» det trenger å fungere.»delta F508 CFTR er fortsatt handikappet, men det er bedre enn ingenting,» sier Guggino. «Og dette betyr at vi ikke bare behandler symptomer lenger. Vi behandler de grunnleggende årsakene.»
Ingen Barn Igjen
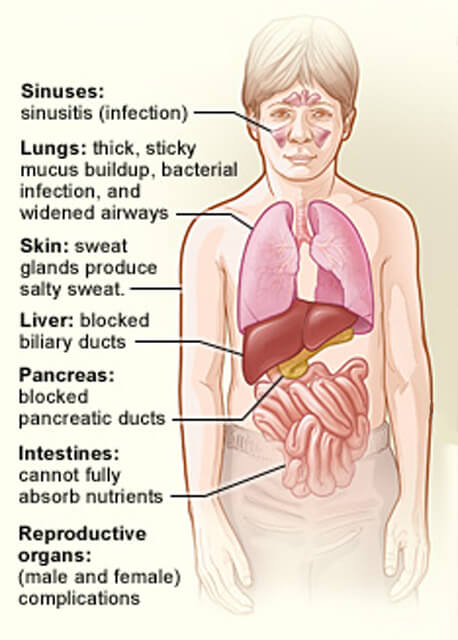 organene som er berørt av cystisk fibrose.
organene som er berørt av cystisk fibrose.Kreditt: National Heart, Lung And Blood Institute
de mer enn 40 prosent av pasientene MED CF med andre mutasjoner-noen ganske sjeldne-er ikke alltid like heldige. DET er over 1700 mutasjoner i CFTR-genet som forårsaker CF, og bare en mindre brøkdel har blitt testet for respons på de godkjente legemidlene. Noen av de testede reagerer på det ene eller det andre stoffet, men andre gjør det ikke. likevel håper forskerne at de en dag vil kunne hjelpe alle. Cutting forutser slutt kategorisere hver pasient ved theratype, et ord han laget for å beskrive grupper av pasienter som er sannsynlig å svare på den samme behandlingen på grunn av en felles underliggende årsak til sine symptomer. Han har jobbet tett med kolleger Patrick Sosnay og Karen Raraigh å utvinne data fra 88,000 personer over hele verden for å teste sitt konsept.Vi kan bruke informasjon generert av eksperimenter i celler til å gruppere mutasjoner som påvirker DEN samme EGENSKAPEN TIL CFTR og derfor bør svare på det samme panelet AV CFTR-legemidler. Gruppering av mutasjoner i henhold til theratype vil muliggjøre kliniske studier på pasienter som bærer forskjellige mutasjoner, i stedet for kliniske studier som evaluerer en mutasjon om gangen,» Sier Cutting. «Det er presisjonsmedisin som blir en realitet .»Dessverre vil stoffbaserte terapier ikke fungere for alle pasienter med CF, spesielt de 2 prosentene som ikke GJØR CFTR i det hele tatt. For disse utvikler forskere måter å målrette mot det muterte genet selv, selv om det fortsatt er mange hindringer å overvinne.Guggino har utarbeidet et genterapisystem som bruker et modifisert adenoassosiert virus (AAV) for å deponere en god versjon AV CFTR-genet inne i cellene. Systemet har vist seg i humane luftveisceller og gnagere.Prøver En annen tilnærming, utviklet Liudmila Cebotaru, Fra Institutt For Medisin, en ny måte å kombinere genterapi og proteinreparasjon ved hjelp av en mekanisme som kalles transkomplementering. I stedet for å plassere CFTR-genet i FULL lengde i AAV, bruker hun en kortere versjon som lettere settes inn i cellens genom. Når det kortere proteinet produseres, binder det seg til pasientens mutantprotein og hjelper det å komme til plasmamembranen. Både Cebotaru Og Guggino tester nå sin nye tilnærming i rhesusaper fordi lungene og immunforsvaret er svært nær mennesker. De håper å starte en klinisk prøve de neste årene, hvis alt går bra.»jeg liker å tenke på det som å hoppe på bilens batteri,» sier hun. «Med litt ekstra hjelp kan PASIENTENS CFTR-proteiner komme til bestemmelsesstedet.»
for både forskere og pasienter er målet intet mindre enn en kur FOR CF. Og selv om det fortsatt er langt unna, er det oppmuntrende tegn på at vi kan komme dit.