de Catherine Gara
ianuarie 2016—mucusul poate să nu fie ceva la care ne place să ne gândim, dar viața noastră depinde de asta. În fibroza chistică (CF), îngroșarea mucusului duce la infecții pulmonare și blocuri intestinale, printre alte simptome. Boala este cauzată de o singură genă afectată de una sau mai multe dintre cele peste 1.700 de mutații despre care se știe că provoacă tulburarea. La Johns Hopkins, cercetătorii și geneticienii clinicieni lucrează împreună pentru a afla tot ce pot despre gena, proteina ei și ce nu merge bine la pacienți, știind că fiecare informație pe care o adună îi aduce mai aproape de a avea opțiuni de tratament pentru toți indivizii cu CF.
Dă-mi sarea
în aceste zile, pacienții cu CF sunt mult mai bine datorită tratamentelor care gestionează infecțiile pulmonare frecvente, dar un diagnostic de CF înseamnă încă o speranță de viață de doar 38 Din cauza taxei pe care o are asupra pancreasului, ficatului și intestinelor. Problema provine din defectele genei regulatorului de conductanță transmembranară a fibrozei chistice (CFTR), care poartă modelul pentru proteina CFTR. Proteina CFTR este ca slotul de poștă într-o ușă din față. Formează un mic pasaj între interiorul și exteriorul celulei.
când funcționează corect, CFTR ajută la controlul trecerii ionilor de clorură (o componentă a sării) în și din celulă. În plămâni și canalele pancreasului, când clorura părăsește celulele, încurajează apa să urmeze. Această apă ajută la formarea straturilor subțiri de mucus. În plămâni, mucusul captează praful și bacteriile care nu ar trebui să fie acolo. Cilia, sau structuri asemănătoare părului, de pe celulele care aliniază plămânii, apoi păstoresc mucusul până la căile respiratorii până la gură, unde este înghițit și trimis pentru a fi digerat. În pancreas, lichidul ajută la transportul enzimelor în intestin pentru a ajuta la digestia alimentelor. Dacă mucusul este prea vâscos în plămâni, cilia nu îl poate muta, astfel încât bacteriile rămân acolo pentru a provoca infecții; dacă este prea vâscos în canalele pancreatice, enzimele nu ajung în intestin, iar alimentele nu se digeră corect.Garry Cutting, profesor la Institutul de Medicină genetică, și Bill Guggino, director al Departamentului de fiziologie, au studiat gena CFTR și proteina codificată pentru cea mai mare parte a carierei lor. Interesul lui Cutting provine din îngrijirea unei perechi de frați cu CF în timp ce era rezident la Johns Hopkins. Interesul lui Guggino se întoarce și mai departe—la călătoriile sale de copilărie la mare. S-a întrebat cum ar putea supraviețui peștii în apa sărată și a aflat că răspunsul scurt este: versiunea lor de CFTR.dacă pare dificil să ne imaginăm concentrarea unei întregi cariere pe o singură genă și produsul său proteic, gândiți-vă la ea în schimb ca la un cub Rubik complex format dintr-un lanț de 1.480 de blocuri magnetice (aminoacizi). O mutație a genei CFTR va însemna adesea o schimbare a unuia dintre aminoacizi, care poate afecta dramatic forma finală tridimensională.
unele mutații determină ca proteina să nu fie produsă. Altele permit sintetizarea unei proteine parțiale. Cu cât mai aproape de începutul proteinei apar aceste mutații, cu atât mai rău pentru funcția sa. Alte mutații apar doar în locul greșit și împiedică deschiderea canalului de sare, de exemplu. Alții fac CFTR să se plieze necorespunzător, ceea ce semnalează echipei de control al calității celulei să o ridice și să o recicleze. Și altele nu interferează cu funcția proteinei, dar o împiedică să ajungă la membrana plasmatică unde își face activitatea.
Cutting și eforturile lui Guggino au contribuit la proiectarea a două medicamente CF pe piață: ivacaftor și lumacaftor. Ivacaftorul activează CFTR purtând mutația G551D. Laboratorul lui Cutting a raportat pentru prima dată această mutație în 1990, iar Guggino și Cutting au generat ulterior informații noi despre efectul acestei mutații asupra funcției CFTR și a simptomelor pacienților. Din păcate, mutația G551D se găsește la doar 4% dintre pacienții cu CF. Cu toate acestea, se pare că 50% dintre pacienții cu CF au două copii ale unei mutații diferite (numită delta F508), ceea ce face ca CFTR să fie slab format și trimis în coșul de reciclare al celulei. Lumacaftor împiedică reciclarea acestuia, astfel încât să ajungă la membrana plasmatică. Apoi, ivacaftor îi dă „lovitura” de care are nevoie pentru a funcționa.
„delta F508 CFTR este încă handicapat, dar este mai bine decât nimic”, spune Guggino. „Și asta înseamnă că nu mai tratăm doar simptomele. Tratăm cauzele profunde.”
nici un copil lăsat în urmă
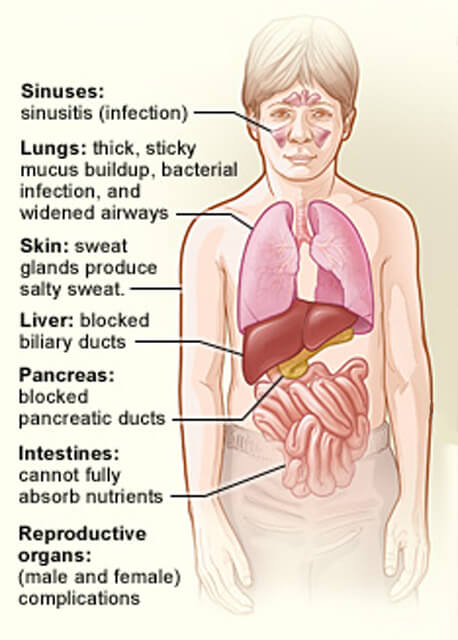 organele afectate de fibroza chistica.
organele afectate de fibroza chistica.Credit: National Heart, Lung și Institutul de sânge
Mai mult de 40 la suta din pacientii cu CF cu alte mutatii — unele destul de rare — nu sunt întotdeauna la fel de norocos. Există peste 1.700 de mutații în gena CFTR care provoacă CF și doar o fracțiune minoră a fost testată pentru răspunsul la medicamentele aprobate. Unii dintre cei testați răspund la unul sau la celălalt medicament, dar alții nu. totuși, cercetătorii speră că într-o zi vor putea ajuta pe toată lumea. Tăierea prevede clasificarea în cele din urmă a fiecărui pacient în funcție de tip, un cuvânt pe care l-a inventat pentru a descrie grupuri de pacienți care sunt susceptibili să răspundă la aceeași terapie din cauza unei cauze comune a simptomelor lor. A lucrat îndeaproape cu colegii Patrick Sosnay și Karen Raraigh pentru a extrage date de la 88.000 de persoane din întreaga lume pentru a-și testa conceptul.
putem folosi informațiile generate de experimente în celule pentru a grupa mutații care afectează aceeași proprietate a CFTR și, prin urmare, ar trebui să răspundă la același grup de medicamente CFTR. Gruparea mutațiilor în funcție de tip ar permite studii clinice pe pacienți care poartă mutații diferite, în loc de studii clinice care evaluează o mutație la un moment dat”, spune Cutting. „Medicina de precizie devine o realitate.”
Din păcate, terapiile pe bază de medicamente nu vor funcționa pentru toți pacienții cu CF, în special pentru cei 2% care nu fac deloc CFTR. Pentru acestea, cercetătorii dezvoltă modalități de a viza gena mutantă în sine, deși există încă multe obstacole de depășit.
Guggino a elaborat un sistem de terapie genică care utilizează un virus adeno-asociat modificat (AAV) pentru a depune o versiune bună a genei CFTR în interiorul celulelor. Sistemul sa dovedit a fi în celulele căilor respiratorii umane și rozătoare.
încercând o abordare diferită, Liudmila Cebotaru, de la Departamentul de Medicină, a conceput o modalitate nouă de a combina terapia genică și repararea proteinelor printr-un mecanism numit transcomplementare. În loc să plaseze gena CFTR de lungime întreagă în AAV, ea folosește o versiune mai scurtă care este mai ușor inserată în genomul celulei. Când se produce proteina mai scurtă, se leagă de proteina mutantă a pacientului și o ajută să ajungă la membrana plasmatică. Atât Cebotaru, cât și Guggino testează acum noua ei abordare la maimuțele rhesus, deoarece plămânii și sistemul lor imunitar sunt foarte apropiați de oameni. Ei speră să înceapă un studiu clinic în următorii câțiva ani, dacă totul merge bine.
„îmi place să mă gândesc la ea ca sărind bateria mașinii tale”, spune ea. „Cu puțin ajutor suplimentar, proteinele CFTR ale pacienților pot ajunge la destinație.”
atât pentru cercetători, cât și pentru pacienți, destinația nu este altceva decât un leac pentru CF. Și, deși este încă un drum lung, există semne încurajatoare că putem ajunge acolo.