Por Catherine Gara
Enero de 2016-El moco puede no ser algo en lo que nos guste pensar, pero nuestras vidas dependen de ello. En la fibrosis quística (FQ), el engrosamiento del moco conduce a infecciones pulmonares y bloqueos intestinales, entre otros síntomas. La enfermedad es causada por un solo gen afectado por una o más de las más de 1700 mutaciones que se sabe causan el trastorno. En Johns Hopkins, investigadores y genetistas clínicos están trabajando juntos para aprender todo lo que puedan sobre el gen, su proteína y lo que va mal en los pacientes, sabiendo que cada pieza de información que obtienen los está acercando a tener opciones de tratamiento para todas las personas con FQ.
Pásame la sal
En estos días, los pacientes con FQ están mucho mejor gracias a los tratamientos que manejan sus infecciones pulmonares frecuentes, pero un diagnóstico de FQ todavía significa una esperanza de vida de solo 38 debido al costo que tiene en el páncreas, el hígado y los intestinos. El problema proviene de defectos en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés), que lleva el modelo de la proteína CFTR. La proteína CFTR es como la ranura de correo en una puerta principal. Forma un pequeño pasadizo entre el interior y el exterior de la célula.
Cuando funciona correctamente, el CFTR ayuda a controlar el paso de iones cloruro (un componente de la sal) dentro y fuera de la célula. En los pulmones y los conductos del páncreas, cuando el cloruro sale de las células, estimula a que siga el agua. Esa agua ayuda a formar capas finas de moco. En los pulmones, el moco atrapa polvo y bacterias que no deberían estar allí. Los cilios, o estructuras en forma de pelo, en las células que recubren los pulmones, luego conducen el moco por las vías respiratorias hasta la boca, donde se traga y se envía para ser digerido. En el páncreas, el líquido ayuda a transportar enzimas al intestino para ayudar en la digestión de los alimentos. Si el moco es demasiado viscoso en los pulmones, los cilios no pueden sacarlo, por lo que las bacterias permanecen allí para causar infecciones; si es demasiado viscoso en los conductos pancreáticos, las enzimas no llegan al intestino y los alimentos no se digieren adecuadamente.
La trayectoria de una proteína
Garry Cutting, profesor del Instituto de Medicina Genética, y Bill Guggino, director del Departamento de Fisiología, han estado estudiando el gen CFTR y su proteína codificada durante la mayor parte de sus carreras. El interés de Cutting proviene del cuidado de un par de hermanos con FQ mientras residía en Johns Hopkins. El interés de Guggino se remonta aún más atrás, a sus viajes de infancia al mar. Se preguntó cómo podrían sobrevivir los peces en agua salada, y se enteró de que la respuesta corta es: su versión de CFTR.
Si parece difícil imaginar centrar toda una carrera en un solo gen y su producto proteico, piense en ello como un cubo de Rubik complejo hecho de una cadena de 1.480 bloques magnéticos (aminoácidos). Una mutación en el gen CFTR a menudo significará un cambio en uno de los aminoácidos, que puede afectar dramáticamente la forma tridimensional final.
Algunas mutaciones hacen que la proteína no se produzca. Otros permiten sintetizar una proteína parcial. Cuanto más cerca del comienzo de la proteína se producen esas mutaciones, peor será su función. Otras mutaciones ocurren en el lugar equivocado e impiden que el canal de sal se abra, por ejemplo. Otros hacen que CFTR se pliegue incorrectamente, lo que indica al equipo de control de calidad de la célula que la recoja y la recicle. Y otros no interfieren con la función de la proteína, pero evitan que llegue a la membrana plasmática donde hace su trabajo.
Los esfuerzos de Cutting y Guggino han contribuido al diseño de dos fármacos para la FQ en el mercado: ivacaftor y lumacaftor. Ivacaftor activa CFTR con la mutación G551D. El laboratorio de Cutting informó por primera vez de esta mutación en 1990, y Guggino y Cutting posteriormente generaron información novedosa sobre el efecto de esta mutación en la función CFTR y los síntomas de los pacientes. Desafortunadamente, la mutación G551D se encuentra en solo el 4 por ciento de los pacientes con FQ. Sin embargo, resulta que el 50 por ciento de los pacientes con FQ tienen dos copias de una mutación diferente (llamada delta F508), lo que hace que la CFTR se forme mal y se envíe al contenedor de reciclaje de la célula. Lumacaftor evita que se recicle para que llegue a la membrana plasmática. Luego, ivacaftor le da la «patada» que necesita para funcionar.
«El delta F508 CFTR sigue siendo discapacitado, pero es mejor que nada», dice Guggino. «Y esto significa que ya no solo estamos tratando los síntomas. Estamos tratando las causas profundas.»
No Child Left Behind
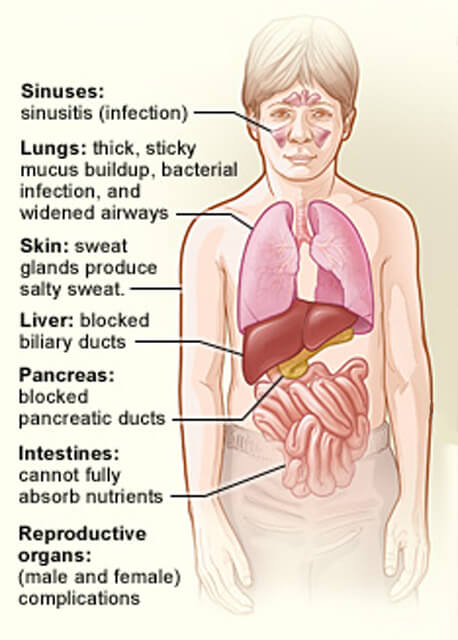 Los órganos afectados por la fibrosis quística.
Los órganos afectados por la fibrosis quística.Crédito: Instituto Nacional del Corazón, los Pulmones y la Sangre
Más del 40 por ciento de los pacientes con FQ con otras mutaciones, algunas bastante raras, no siempre son tan afortunados. Hay más de 1,700 mutaciones en el gen CFTR que causan la FQ, y solo una fracción menor se ha probado para determinar la respuesta a los medicamentos aprobados. Algunos de los examinados responden a uno u otro medicamento, pero otros no. Sin embargo, los investigadores tienen la esperanza de que algún día podrán ayudar a todos. Cutting prevé finalmente categorizar a cada paciente por el tipo, una palabra que acuñó para describir grupos de pacientes que probablemente responderán a la misma terapia debido a una causa subyacente común de sus síntomas. Ha estado trabajando estrechamente con sus colegas Patrick Sosnay y Karen Raraigh para extraer datos de 88,000 personas en todo el mundo para probar su concepto.
Podemos utilizar la información generada por experimentos en células para agrupar mutaciones que afectan la misma propiedad de CFTR y, por lo tanto, deben responder al mismo grupo de fármacos CFTR. Agrupar mutaciones de acuerdo con el tipo de mutación permitiría realizar ensayos clínicos en pacientes con diferentes mutaciones, en lugar de ensayos clínicos que evalúan una mutación a la vez», dice Cutting. «Eso es que la medicina de precisión se está convirtiendo en una realidad.»
Desafortunadamente, las terapias basadas en medicamentos no funcionarán para todos los pacientes con FQ, especialmente para el 2 por ciento que no realiza CFTR en absoluto. Para estos, los investigadores están desarrollando formas de atacar el gen mutado en sí, aunque todavía hay muchos obstáculos que superar.
Guggino ha desarrollado un sistema de terapia génica que utiliza un virus adenoasociado modificado (AAV) para depositar una buena versión del gen CFTR dentro de las células. El sistema ha demostrado su eficacia en células de vías respiratorias humanas y roedores.
Probando un enfoque diferente, Liudmila Cebotaru, del Departamento de Medicina, ideó una forma novedosa de combinar la terapia génica y la reparación de proteínas mediante un mecanismo llamado transcomplementación. En lugar de colocar el gen CFTR de longitud completa dentro de AAV, está utilizando una versión más corta que se inserta más fácilmente en el genoma de la célula. Cuando se produce la proteína más corta, se une a la proteína mutada del paciente y la ayuda a llegar a la membrana plasmática. Tanto Cebotaru como Guggino están probando su nuevo enfoque en monos rhesus porque sus pulmones y sistemas inmunológicos están muy cerca de los humanos. Esperan comenzar un ensayo clínico en los próximos años, si todo va bien.
«Me gusta pensar que es como saltar la batería de tu auto», dice. «Con un poco de ayuda adicional, las proteínas CFTR de los pacientes pueden llegar a su destino.»
Tanto para los investigadores como para los pacientes, el destino es nada menos que una cura para la FQ. Y aunque todavía está muy lejos, hay señales alentadoras de que podemos llegar allí.